
2023年3月23日下午,线上报告——“反应堆材料的第一性原理模拟计算”已通过源资课堂顺利举办。
本次报告中,中山大学刘文冠副教授介绍了借助第一性原理、分子动力学、机器学习力场等方法研究核材料腐蚀和氢同位素等问题,并且提出理论模拟对于反应堆难以开展相关实验的极端条件已经成为了一种必不可少的研究手段。
报告主要内容
铅铋腐蚀的模拟研究
熔盐堆合金的晶界腐蚀研究
镍基合金中合金元素对氢同位素的影响
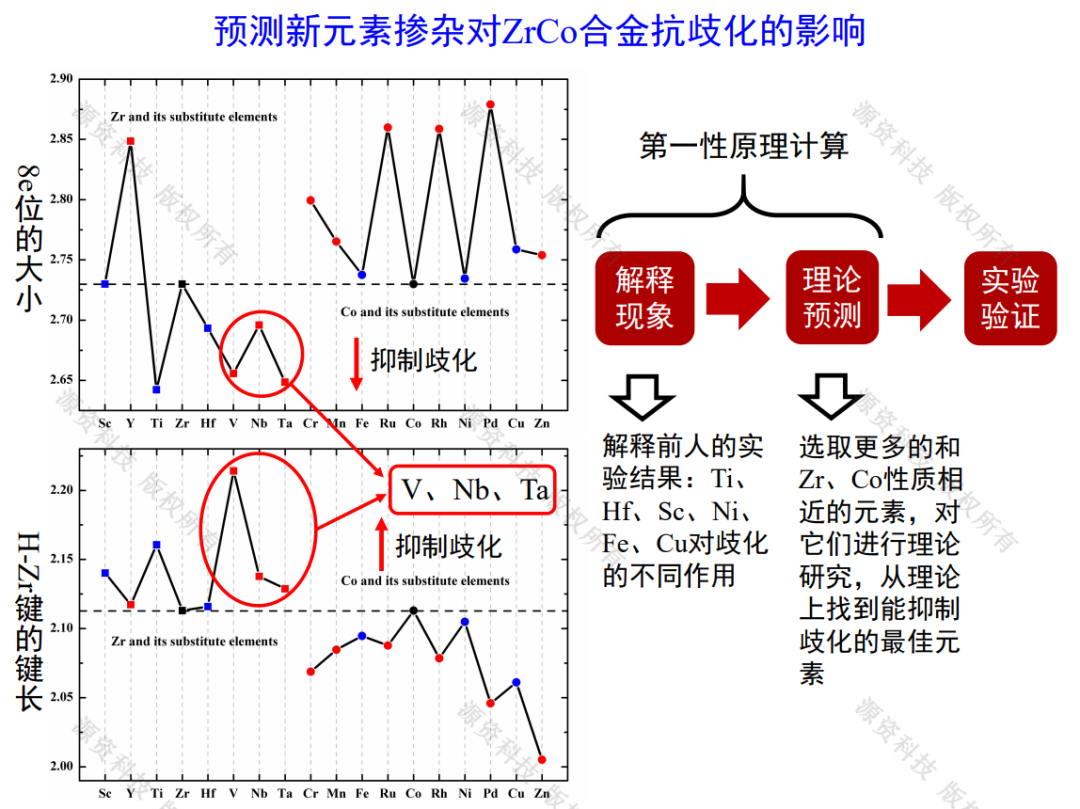
ZrCo系储氚合金歧化机理及改良研究
核石墨中氢同位素吸附/脱附问题的研究
熔盐堆中氚分布的理论计算
孪晶强韧化等的理论模拟
一些相关熔盐的热物性相图研究
课堂精彩内容分享



Q&A答疑分享
Q1:在熔盐相图的计算中,通过第一性原理计算的基础数据都包括哪些呢?
主要的基础数据包括各个组分的形成能。如果有组分有多种晶体结构的话,需要找到形成能最低的那个结构。
Q2:“最优电荷密度面”应该怎么理解?
氢原子在合金中倾向存在于特定电荷密度处,也就是“最优电荷密度面”,并且不同的金属中的最优电荷密度值不同。
Q3: 关于合金的建模,如何处理溶质元素比较好?
合金中有替换位置和间隙位置,需要根据所处位置的结合能最低来判断。通常金属元素占据替换位置,半径较小的非金属元素占据间隙位置。
Q4:模拟过程中氕氘氚是否都可以统一按氢元素来处理?
氢同位素的核外电子排布可以认为几乎没有差别,所以研究电荷密度或者反应势垒时,可以忽略它们直接的差别。但是分子动力学模拟或者计算扩散系数,就需要考虑原子质量,三者的质量是不一致的,必须区分。
Q5:如何使用VASP进行高熵合金涂层耐铅铋腐蚀计算?有哪些难点?
首先要对高熵合金进行建模,可以使用MedeA中的SQS(准随机近似)功能。高熵合金计算麻烦的地方在于,需要计算一系列值,然后平均值,导致计算量很大。
Q6:铅铋块堆防腐蚀涂层的选材怎么通过第一性原理进行筛选?
耐铅铋腐蚀涂层的选材很复杂,需要考虑很多因素。第一性原理计算通常只能定性地给出一些建议,比如通过计算合金元素的结合能来判断其耐溶解腐蚀的能力。
报告回看方式
扫描上方二维码,关注“源资科技VASP”公众号

回复“反应堆”,点击收到的链接即可进入直播间免费观看报告回看


