
前言:
鸟枪宏基因组学能够比较全面的进行微生物的分型和病原体的鉴定。但是,在实际操作的过程中,还有非常多的因素影响鉴定的准确性。本文探讨了鸟枪法宏基因组分析用到的工具,其中CLC Genomics Workbench在后期生信分析中能提供更为准确的结果。
摘要:
应用于宏基因组研究的高通量测序能为临床的患者样本进行诊断或分子分型提供的一站式解决方案,能够快速有效的提出适合疾病的治疗方案或针对某一传染病提供预防和监控措施。然而,由于方法的多样,目前还不清楚各种可用的方法如何影响最终结果。在本文中,我们用鸟枪法宏基因组处理了多种来源的患者样本,并且在提取宏基因组DNA之前,使用三种不同的方法消除人类DNA。并且采用临床微生物实验室中常用的基因组学数据分析方法进行分析。数据分析的结果与传统的微生物病原体培养的方法进行比较。结果,75%的测序reads都来源于人类DNA,是分析的主要干扰因素,没有一种试剂盒能够有效的降低宿主与微生物DNA的初始比例。而用于微生物分型及抗生素耐药性研究的生物信息学工具对比结果尽管受到微生物DNA测序深度和采用的数据库的限制,CLC Genomics Workbench呈现的结果还是明显优于其他的分析工具。
参考文献:
Couto N, Schuele L,Rossen JW, et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Scientific Reports, 2018 Sep 13;8(1):13767.
试验方法:
1、 采用9种患者体液和1种组织样本进行宏基因组的测序研究。
2、样本分为三组,用不同的DNA提取方法进行DNA的提取,测序。
3、数据分析结果用不同的生物信息学软件进行分析,并与传统细菌培养结果进行对比。
部分对比结果:

表一. 样本类型及处理方法。测序结果采用CLC Genomics Workbench v10.0.1进行reads的hg19的mapping。
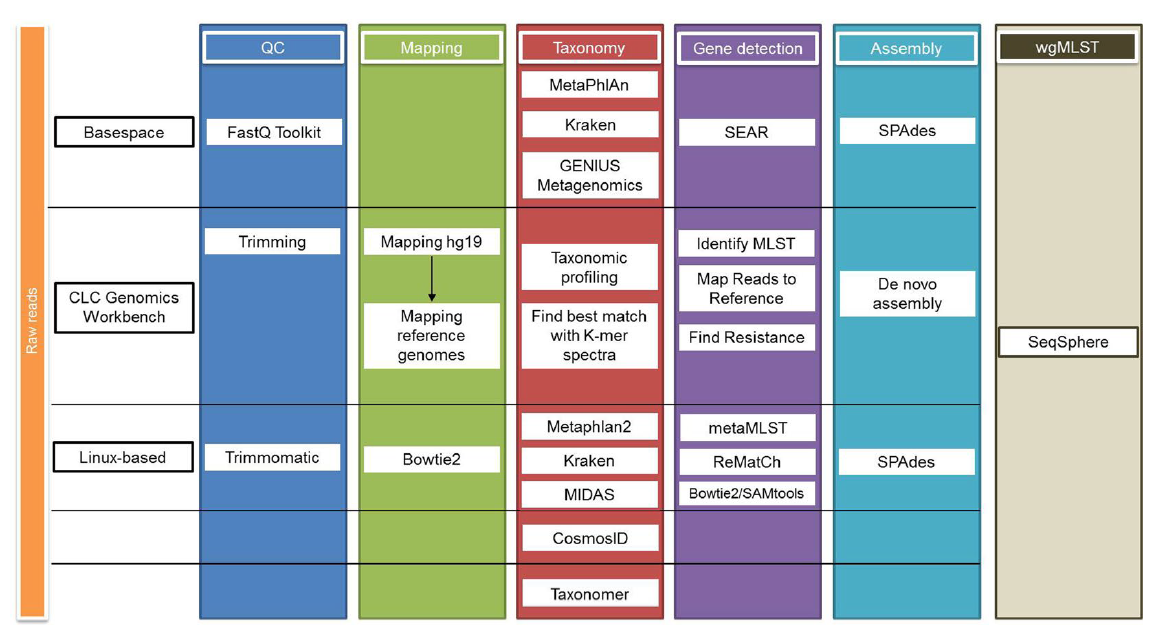
图一. 数据分析采用的三个平台及其数据分析所用的步骤和算法。

表二. 采用网页分析平台进行Shotgun metagenomics的数据分析结果。对比了传统的细菌培养方法、基于WGS鉴定的方法和鸟枪法宏基因组学的方法。Kraken使用总的reads数作为分母计算相对丰度。剩下两个方法采用classified reads作为分母计算相对丰度。#代表尽管实验室培养能够鉴定病原体,但没有分离物用于WGS。*代表没有匹配的病原体。

表三:采用CLC genomics Workbench平台进行Shotgun metagenomics的数据分析结果。CLC genomics workbench采用classified reads作为分母计算相对丰度。#代表尽管实验室培养能够鉴定病原体,但没有分离物用于WGS。*代表没有匹配的病原体。

表四. 采用依赖Linux平台进行进行Shotgun metagenomics的数据分析结果。b使用总的reads数作为分母计算相对丰度。c采用classified reads作为分母计算相对丰度。#代表尽管实验室培养能够鉴定病原体,但没有分离物用于WGS。*代表没有匹配的病原体。

表五. 每个样品的不同分类方法的性能。以培养/MALDI-TOF为标准计算灵敏度和阳性预测值。PPV,阳性预测值。
病原体检测对比结论:
● 宏基因分析方法仅需要48-54个小时,而传统的分析方法往往需要1-2周才能鉴定。
● 本文采用了三种有代表性的的宏基因组分析工具进行对比:一、基于网页的分析平台(收费/免费);二、基于Unix分析软件;三、全视窗化操作商业分析软件(CLC genomics workbench)。表五可见:如果采用基于Unix的分析软件,MetaPhlAn2是一个不错的选择,在比较灵敏的同时保证了较佳的阳性预测值。如果选用全视窗化的CLC Genomics Workbench,其提供的Best match K-mer spectra工具也具有较佳的阳性预测值。
● 本文测试三个有代表性的平台多种做宏基因组的pepline,CLC Genomics Workbench比其他方法要有很多优势,其不仅能够提供较准确的分析结果,而且具有非常友好的用户界面。不需要具有Unix背景就能够快速搭建宏基因组分析平台。相较于网页平台,CLC提供了更全面的工具可以选择。
● 虽然宏基因组分析的方法目前还不能完全替代实验室培养,但是随着数据库的不断完善,相信宏基因组的分析方法能够很快为医疗诊断和预防提供有效的工具。
*本文还对比了不同平台对样本中细菌抗生素耐药性的分析结果,篇幅有限,可查看原文获得相关结果。


