
有机材料密度、内聚能、汽化热的分子动力学研究
1. 研究背景
高分子有机材料广泛应用在我们生活中的各个领域,比如玻璃,塑料,胶水等,其形态和性质也会各不相同。因此,能够准确预测有机材料的各种理化性质一直受到关注。其中,简单烃类化合物常被作为溶剂、燃料等方面应用,实验数据较为完善,因此特别适合用于验证模拟方法的可靠性。模拟技术经验表明,烃类化合物的模拟准确度在一定程度上也代表了相关有机体系的准确度。
分子动力学基于牛顿力学的计算原理,计算各原子的受力后,对牛顿方程进行积分,从而得到体系性质随时间变化的过程,是基于原子尺度的模拟计算。在分子动力学中,用于计算体系能量和力的函数形式称为力场,力场的精度决定了性质模拟的准确度。采用合适的力场,分子动力学模拟就能够得到高精度的结果。通常来说,模拟与实验的结果吻合度相当高,常在1% 以内。现代的有机物力场几乎覆盖了所有常见分子体系,并具有高度的适用性。本案例以直链烃化合物为例,详细介绍了的有机液体模型构建、分子动力学计算过程以及详细的结果分析。
2. 有机材料模型
在本案例中,作者通过MedeA-Polymer Builder建模工具,从标准重复单元结构数据库中选择了烃类化合物单体(图1(a))。然后,通过MedeA-Amorphous Materials Builder建模工具创建了凝聚态高分子模型(图1(b))。

图1 (a) 烃类化合物单体结构(b)聚合物模型结构
3. 选择力场
采用MedeA-LAMMPS提供的力场,可以确定高分子聚合物的原子类型和电荷类型。其中,力场原子类型可以根据标准模板自动指定。MedeA-LAMMPS免费提供了多种广泛使用的力场。本案例使用的适用于有机烃类物质的PCFF+力场是在PCFF力场基础上进行了精修,所用方法与早期开发的COMPASS力场类似。
4. 模拟过程以及计算参数选择
通过MedeA-Polymer Builder构造出的正己烷聚合物的模型,可以用MedeA-LAMMPS进行NPT系综下的分子动力学计算。在计算过程中,作者评估了不同性质计算对模型大小和模拟时长的敏感性,然后再确定了优质模拟体系大小和时长。图2中,MedeA提供的Flowchart流程图功能,极大地方便了分子动力学模拟的过程。随后,作者使用相同方法研究从正戊烷到正二十烷所有直链烃的分子动力学过程。

图2. MedeA-LAMMPS 模拟凝聚相烃类化合物的流程图
靠前NPT步骤在100 ps时间内,外压从高压减小至设定压力P2,第二个NPT步骤则将压力恒定在P2 = 1 atm下
图3给出了正己烷密度随体系大小(a)和模拟时长(b)的变化关系,不确定度可以由模拟过程的涨落给出。很明显,对于正己烷来说,包含216个分子的模型和200ps的时长能够很好地描述体系的密度这一性质。经验表明,对于绝大多数体系来说,3500个原子的体系大小与200ps的NPT时长可以给出足够好的结果。实验上正己烷在298.15K下的密度是0.6548g/cm3,模拟结果得到0.6547g/cm3,模拟值仅比实验值低0.02%。
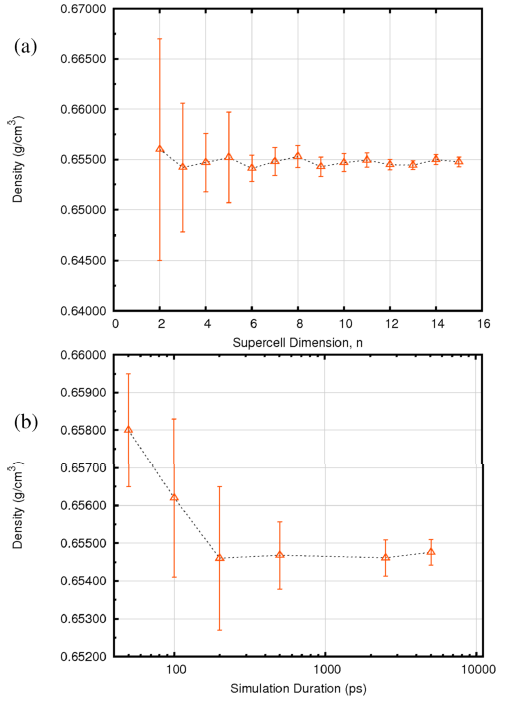
图 3正己烷密度的计算结果(25°C,1 atm)随(a)体系大小(分子数为n3)(b)模拟时长(ps) 对于正己烷,216个分子(n=6)、200ps时长即可得到收敛结果
5. 烃类性质计算
采用MedeA-LAMMPS的流程图方法可以计算气体、液体、固体的分子动力学过程。MedeA中的流程图可保存,可重复使用,这便于将一套完整、类似的模拟过程应用与不同的体系。表1为不同碳数的烷烃在25°C、1atm下的密度(ρ)、内聚能密度(CED)和气化热数值(ΔHvap)。 PCFF+力场给出的密度数据相对实验值的偏差在0.5%以内;表1中也列出了内聚能密度(CED),实验上的气化热与内聚能密度密切相关,使用PCFF+力场的计算数据与实验值偏差小于1%。 PCFF+给出密度和气化热的平均绝对误差分别为0.23%和0.28%,使用PCFF+比COMPASS力场得到了更准确的结果。
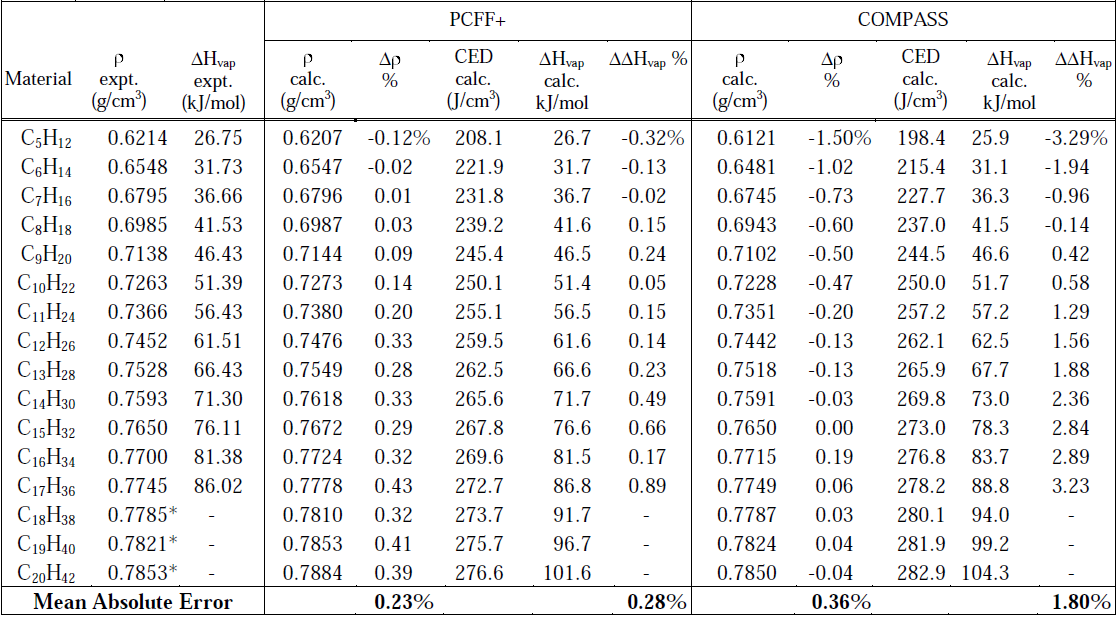
表1 计算得到的烷烃在25°C、1atm下的密度(ρ)、内聚能密度和气化热。
Δρ%和ΔΔHvap%为实验和计算值的偏差。
表1的结果显示烃类化合物的性质计算可以得到与实验数值吻合的密度、内聚能密度和气化热。很多其他理化性质例如热导率、粘度、密度随压力的变化函数等也可以得到,而且模拟得到的结果与实验符合得很好。此外,小分子(少于15个碳原子)扩散性质也可以用类似方法得到,相应的自扩散系数可以直接用分子动力学方法给出。分子模拟在材料性质预测上的成功结果有效地帮助了实验上对各种材料的初步虚拟筛选,从而加快实验上的材料合成。
6. 总结与展望
通过分子动力学,可以有效地进行高分子有机材料的性质模拟。分子动力学过程轨迹通常在纳秒尺度,因此常常用于模拟缓慢的动态过程,例如高分子体系以及大分子体系的扩散等,难度较大,而且研究体系的大小也通常限制在几千个原子之内。然而,随着计算性能的提升,对于体系尺度的限制也在被逐步打破。MedeA-LAMMPS可以很方便的进行大规模的并行计算,随着超级计算机的发展,分子动力学模拟的时间尺度逐渐进入微秒级别,体系大小也进入上百万个乃至几十亿个原子。事实上,只要采用合理的方法和足够的计算资源,分子动力学在预测与研究有机材料的性质等方面将会起到越来越重要的作用!
参考文献:
H. Sun, S. J. Mumby, J.R. Maple, A. T. Hagler, J. Phys. Chem. 99, 5873 (1995)
H. Sun and D. Rigby, Spectrochimica Acta A153, 1301 (1997)
D. Rigby, H. Sun, B.E. Eichinger, Polymer International 44, 311 (1997)
H. Sun, J. Phys. Chem. B102, 7338 (1998)
H. Sun, P. Ren, J.R. Fried, Comput. Theor. Polymer Sci. 8, 229 (1998)
S. Plimpton J. Comp. Phys. 117, 1 (1995); LAMMPS官网: http://lammps.sandia.gov/
F. Abraham., R. Walkup., H. Gao., M. Duchaineau., T. D. De La Rubia., M. Seager. Proc. Natl. Acad. Sci. 99, 5783 (2002)
M.L. Klein, W. Shinoda, Science 321, 798 (2008)
使用MedeA模块:
Welcome to MedeA Bundle
MedeA-Amorphous Materials Builder
MedeA-LAMMPS
MedeA-LAMMPS-CED


