
SiC相转变研究
1. 研究背景
众所周知,密集排列的层状晶体化合物如SiC、ZnS、CdI2、MoS2等,按交替原子层,有大量相异的相位。例如SiC有100多个相,每个中原子能量几乎相同。立方型SiC-3C带隙为2.4eV,六方型SiC-4H带隙为3.3eV,电子迁移率在这些构型中比较高,基于异质结3C/4H的设备有比较大的应用前景。本案例中,作者采用密度泛函理论方法,重点研究了2Hà 6H和3Cà 6H转变,计算转变能量分布及过渡态。
2. 建模与计算方法
作者通过Welcome to MedeA Bundle中InfoMaticA搜索了SiC结构,随后创建了SiC-2H、SiC-3C及SiC-6H不同体系,接着采用MedeA-VASP模块中采用密度泛函DFT的方法,对各体系进行结构优化;K点采用7x7x3,截断能为400eV,采用MedeA-TSS模块搜索2Hà 6H和3Cà 6H转变反应路径。
3. 结果与讨论
3.1 SiC结构优化
作者采用MedeA-VASP模块优化SiC-2H、SiC-6H结构,见图1。图1(a)中,对于SiC-2H,SiC密集填充层,垂直方向上,从A位置移动到C位置;图1(b)中,从位置B移动到位置C。SiC-3C结构,见图2。转变位点为A和B。


图1 SiC-2H转变为SiC-6H各结构
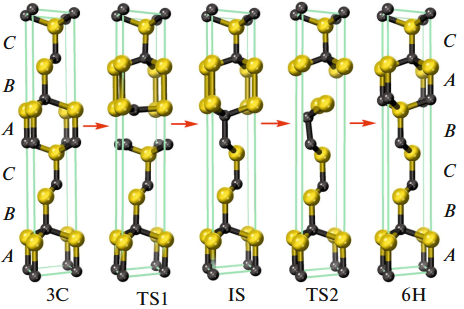
图2 SiC-3C转变为SiC-6H各结构
3.2 2Hà 6H及3Cà 6H相转变计算
随后,作者采用MedeA-TSS模块计算了2Hà6H相转变过程,见图3,。2Hà6H转变中有三个极小值,中间最小值(中间IS)是由于两个链接位点改变形成,位点改变不是同时进行,而是轮流改变。当第一层中A迁移到C,B才相C迁移,见图1。除此之外,还有一种可能,B先向C迁移,然后再是A向C迁移。中间态IS对称性P3m1,比末态、初态对称性P63mc能量低。NEB计算显示,从2HàIS,ISà 6H分别有一个中间亚稳态IS1和IS2,亚稳态对称性为Cm。

图3 SiC-2H转变为SiC-6H反应路径
作者进一步研究了3Cà6H转变过程,见图4。3Cà6H转变中需要Si-C键断裂,形成新的Si-Si键和C-C键,然后它们断裂,最后形成新的Si-C链。SiC中Si-Si和C-C键形成需要大量能量,因此,转变能量明显高于2Hà6H能垒,能量为350 KJ/mol (3.6eV/atom)。3Cà 6H转变中,IS对称性为P3m1,低于初态(对称性为F43m)和末态(对称性P63mc)。反应过程中TS1和TS2对称性为Cm。

图4 SiC-3C转变为SiC-6H反应路径
4. 结论
综上,作者通过第一性原理计算研究表明在2Hà6H中,通过SiC键压缩实现转变,而3Cà6H中,通过Si-Si和C-C键辅助完成转变。因此2Hà6H (1.7eV/atm) 能垒明显小于3Cà6H (3.6eV/atm),通过分析过渡态发现,所有过渡态结构均具有单斜对称性。通过此次研究,有助于我们更好理解SiC相转变性质,为我们日后对碳化物更深层的次研究做出较大贡献。
参考文献:
S.A. Kukushkin, A.V. Osipov. Thechniques for Polytypic Transformations in Silicon Carbide. Physics of the Solid State. 2019, Vol. 61, No. 8, pp, 1389-1393.
使用MedeA模块:
Welcom to MedeA Bundle
MedeA-VASP
MedeA-TSS


