
氧溶液和Mo/Cr界面偏析效应的第一性原理计算
Mo基体-Cr涂层界面、掺杂O原子、裂纹、抗腐蚀
一、案例背景
自2011年福岛灾难以来,为了提高核反应堆的安全性和效率,人们对于在非正常条件下能更好地抵御氧化的轻水反应堆(LWR)覆层材料的开发表现出了极大兴趣。钼、铌和钽等耐火合金是一种潜在的抗事故覆层材料。然而,钼的抗氧化性研究在LWR中是一个重要问题。本案例作者使用第一性原理计算方法评价了Mo/Cr界面的性能,并通过优化界面微观结构,加深对Mo/Cr键合性能的了解,为指导微界面抗氧化行为,研究在核包覆材料科学领域的应用提供了方向。
二、建模与计算方法
作者通过MedeA InfoMaticA 搜索了Mo和Cr晶胞。随后将O原子分别掺杂至Mo和Cr晶胞的间隙位置和取代位置,创建了掺杂模型;利用Surface Builder切面得到Mo和Cr晶胞(100)和(110)面;随后使用Interface Builder搜索了界面结构,构建了Mo(100)/Cr(100)、Mo(110)/Cr(110)和Mo(110)/Cr(100)界面模型;最后通过Find empty space在界面模型中加入了O原子,组建了掺杂模型。作者采用MedeA VASP模块中GGA-PBE方法对一系列体系结构进行优化,并计算了溶解能、表面能、功函数、黏着功等等。使用MedeA MT分析了材料的力学性质。体相计算:截断能500 eV,K点:6×6×6;表面计算:截断能500 eV,K点:2×2×1。考虑过渡金属d轨道强耦合作用,采用hubbard U校正(UMo=2.05 eV;UCr=3.2 eV)。
三、结果与讨论
3.1晶体结构和力学性质分析
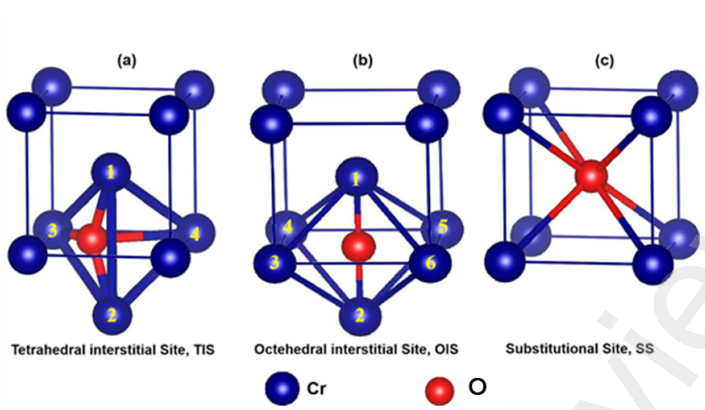
图1 (a) 四面体间隙位 (TIS);(b) 八面体间隙位 (OIS);(c) 取代位 (SS)
表1 Mo和Cr单晶胞的理论和实验弹性常数Cij(GPa)、体模量B(GPa)、剪切模量G(GPa)、杨氏模量E(GPa)、泊松比σ

作者利用MedeA VASP计算方法得出Mo和Cr的优质晶格常数及力学性质,并且结果与实验数据非常吻合。Mo在含有游离氧的环境中易氧化,而Cr涂层可用于保护。在高温下,纯蒸汽分解的氧原子在Mo晶体和Cr晶体中分别更倾向于四面体间隙位点和八面体间隙位点。除氧原子外,Cr和Mo原子也可以相互替换。Cr取代Mo的过程较为容易。
3.2表面模型分析
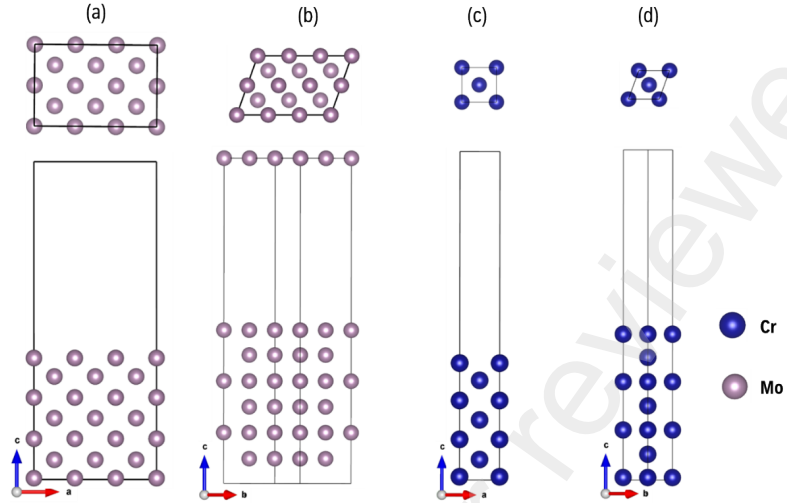
图2 优化后的 (a) Mo(100)面;(b) Mo(110)面;(c) Cr(100)面;(d) Cr(110)面
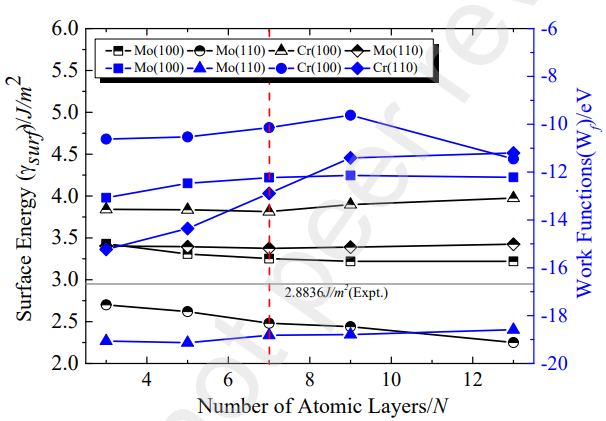
图3 3到13层Mo(100)、Mo(110)、Cr(100)、Cr(110)的表面能和功函数
为了构建Mo/Cr界面,作者首先构建了Mo(100)、Mo(110) 、Cr(100)、Cr(110)表面,如图2所示,并考察了不同原子层数对表面结构的影响。如图3所示,作者计算了从3到13层的各表面的表面能,分析发现,当原子层数为7层时Mo表面和Cr表面都可以展现类似于块体Mo和Cr的特性。为了反映随着原子层数的增加而变化的表面覆盖度,作者计算了不同层数表面的功函数。当各表面原子层数为7层时,能够有效的反映表面特性并平衡计算效率与成本。
3.3界面能和黏着功分析
表2 Mo(100)/Cr(100)、Mo(110)/Cr(110)和 Mo(100)/Cr(110)界面的黏着功和界面能
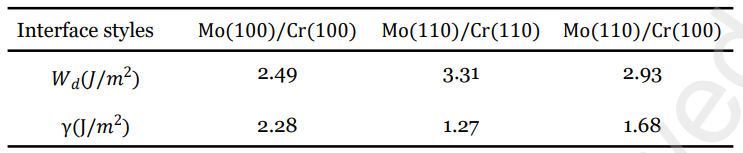
作者考虑了晶格失配率和界面失配的情况,获得了三种稳定的界面结构:Mo(100)/Cr(100)、Mo(110)/Cr(110)和Mo(110)/Cr(100)。其中,Mo(110)/Cr(110)界面表现出最强的界面键合强度,黏着功最大,也具有最小的界面能,因此是最稳定的;而Mo(100)/Cr(100)界面则相对较弱,其界面能最大。
3.4界面局部原子结构分析

图4 (a) Mo(100)/Cr(100)界面;(b) Mo(110)/Cr(110)界面;(c) Mo(110)/Cr(100)界面的局部原子优化前后对比
界面处Mo/Cr的互扩散可能会降低Cr涂层的保护性。通过对结构优化后的三种Mo/Cr界面进行分析,发现Mo和Cr原子在界面处发生了不同程度的重排,导致界面距离的变化。Mo/Cr界面距离大约保持在2.1~2.4Å左右。
3.5含O杂质界面结构分析
为了研究O原子的偏析及其对界面稳定性的影响,作者构建了带有O杂质的5种典型界面结构并进行了计算分析。结果表明,杂质氧原子在界面位置会聚集,并可以增强界面键合强度。Mo易受腐蚀和氧化影响,而Cr作为涂层时可以与O杂质产生稳定的Cr2O3保护层,从而降低Mo的腐蚀率。
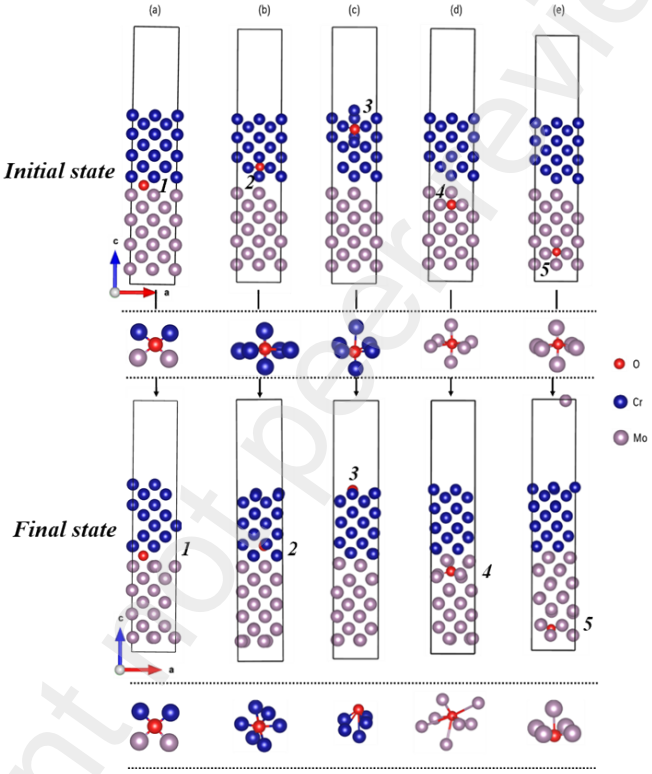
图5 (a) 带有杂质氧原子的 Mo(100)/Cr(100)界面;(b) 带有靠近Cr表面边缘的杂质氧原子的Mo(100)/Cr(100)界面;(c) 带有远离界面处Cr晶格中的杂质氧原子的Mo(100)/Cr(100)界面;(d) 带有靠近Mo表面边缘的杂质氧原子的 Mo(100)/Cr(100)界面结构;(e) 带有远离界面处Mo晶格中的杂质氧原子的Mo(100)/Cr(100)界面
四、总结与展望
本案例使用密度泛函理论,对Mo和Cr体相、表面、界面的多个性质进行了计算和分析,包括结构性质、力学性质、杂质氧原子的溶解性能等。计算结果表明,使用GGA+U方法得出的Mo和Cr的晶格常数、体模量、剪切模量、杨氏模量和Possion比与实验数据吻合;通过界面能和黏着功计算,发现Mo(110)/Cr(110)的界面是最稳定的;而杂质氧原子更倾向于溶解在Cr表面边缘,同时带有杂质氧原子的界面黏着功更高,这意味着杂质氧原子能够增强Cr涂层Mo合金的粘附机制,从而降低Mo的腐蚀率。本研究为Mo和Cr的应用提供了参考价值,并对其多项性质进行了理论计算和分析。
参考文献
http://dx.doi.org/10.2139/ssrn.4003178
✔如需以上案例详细介绍,请点击“阅读原文”即可下载PDF文档。


