
1. 案例背景
碳支撑催化剂在现代化学工业中发挥着至关重要的作用,不同组分之间的界面相互作用最值得注意,因为它被视为影响催化性能的关键因素。人们已经对对碳支撑金属催化剂(金属/碳催化剂)中的金属-支撑相互作用(MSI)进行了全面研究,其中电子金属-支撑相互作用(EMSI)和强金属-支撑相互作用(SMSI)的重要概念已经完全确立。近期,研究者在碳支撑的氧化物催化剂(氧化物/碳催化剂)中也发现了氧化物-支撑相互作用,但具有良好还原性的过渡金属磷化物却鲜有报道。在本项工作中,作者提出了电子磷化物-支撑相互作用(EPSI)的新概念,并通过使用氮磷共掺杂碳(NPC)支撑的磷化钼(MoP)作为模型催化剂(MoP@NPC)证实了这一概念。这种强EPSI不仅能使MoP在环境条件下稳定在低氧化态,还能调节其电子结构,从而降低含氧中间产物的解离能,提高氧化脱硫的催化活性。
2. 建模与计算方法
作者通过MedeA Environment中InfoMaticA数据库搜索MoP结构,采用Surface Builder功能创建MoP(001)表面;采用Molecule Builder功能创建氮磷共掺杂碳(NPC)结构。作者采用MedeA-VASP模块中GGA-PBE方法,对体系进行结构优化并分析能带结构、态密度、电荷密度等电子性质,计算过程考虑范德华作用力(DFT+D3);MoP晶胞优化截断能为520 eV,其余体系设置截断能为400 eV;结构优化K点2x2x1,电子性质K点4x4x1;电子收敛标准为10-5 eV/atom。
3. 结果与讨论
3.1 实验研究
作者以有机-无机杂化结构的钼基金属-有机框架(Mo-MOFs)为Mo前驱体制备了MoP@NPC复合材料,随后通过SEM、TEM、XRD等表征结构,见图1。从图1中可知:
(1)MoP@NPC呈现垂直排列的二维薄片构型(厚度约100 nm),并形成完全互连多孔结构;
(2)TEM图像中纳米级MoP紧密嵌入碳层(图1d);HRTEM中观察到NPC覆盖层与MoP晶体之间存在界面接触(图1e);
(3)XRD证实MoP@NPC中MoP晶体的随机取向和多晶特性(图1f, g);
(4)元素映射图表明,Mo、P、N和C在MoP@NPC复合材料中分布均匀(图1h-k)。综上说明成功制备MoP@NPC复合材料。
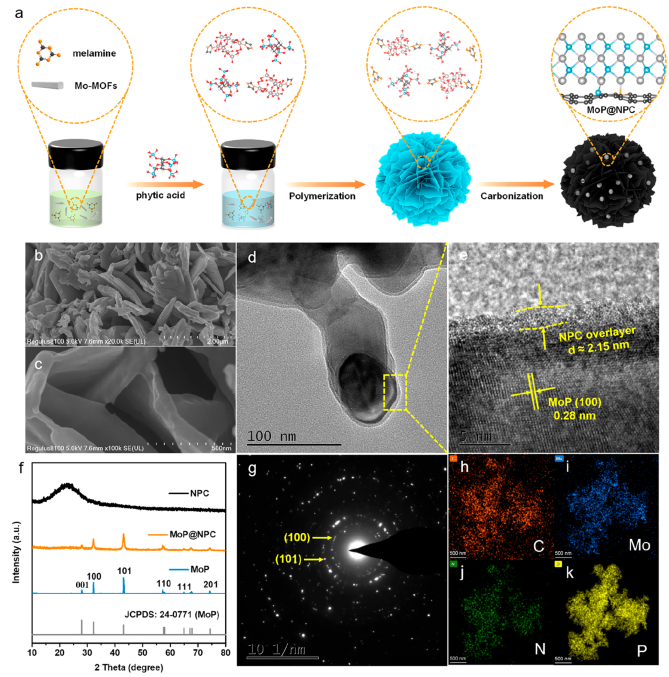
图1 MoP@NPC合成及结构表征:(a) 合成示意图;(b, c) SEM图像;(d) TEM图像;(e) HRTEM图像;(f) XRD图像;(g) SAED图像;(h-k) 元素映像图
随后作者对MoP@NPC样品进行化学性能表征,见图2。从图中可知:MoP@NPC的Electron paramagnetic resonance (EPR)强度远高于MoP,说明MoP@NPC中低价Mo(Mo3+和Mo4+)含量高,说明NPC能显著降低MoP表面氧化,且存在强电子相互作用;XPS与拉曼光谱显示,MoP@NPC中Mo 3d峰移至正的位置(图2b)。上述结果表明电子从MoP向NPC转移,MoP与NPC之间存在强电子相互作用。MoP@NPC中MoP表面保持稳定,不易氧化,这与EMSI中金属-支撑相互作用概念相似,作者提出了氧化物/碳催化剂中的电子氧化物-支撑相互作用(EOSI)概念。
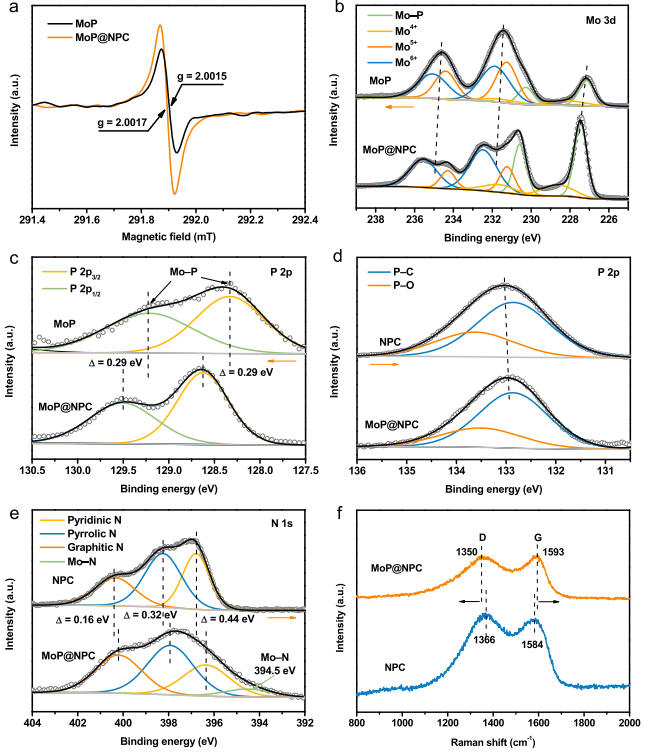
图2 MoP@NPC样品表征:(a) EPR谱图;XPS光谱 (b) Mo 3d,(c, d) P 2p,(e) N 1s;(f) 拉曼光谱
3.2 理论研究
3.2.1 EPSI(电子磷-支撑相互作用)理论研究
作者采用MedeA VASP模块验证MoP@NPC中强的EPSI。通过理论计算发现:
(1)MoP@NPC中界面键能极负(-8.79 eV),表明MoP和NPC之间存在强相互作用;
(2)MoP@NPC的平面静电位(图3a),MoP和NPC界面上有一个巨大的势阱,再次证明界面间有强的电子相互作用。
(3)电荷密度和Bader电荷(图3b)显示,NPC通过界面和化学键从MoP中捕获5.06个电子。理论计算结果与实验结果高度一致,共同揭示了MoP和NPC之间存在界面化学键和强EPSI。
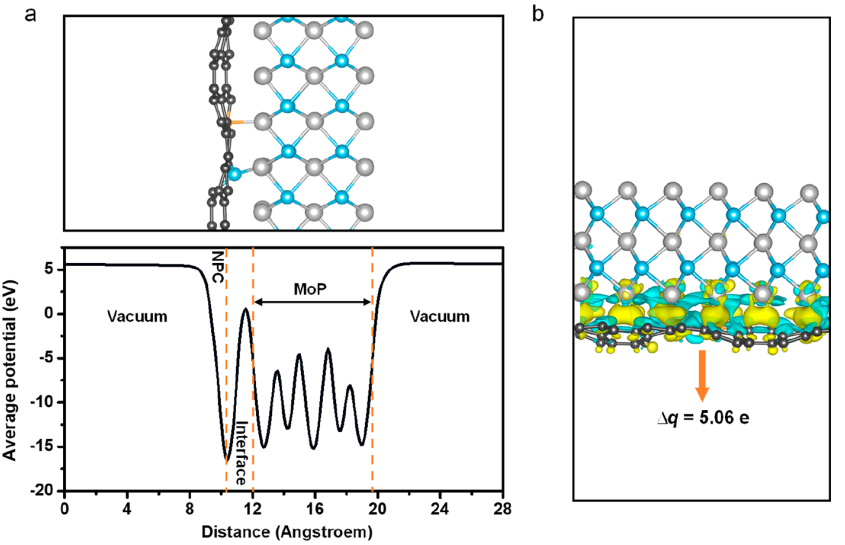
图3 (a) MoP@NPC结构及平均静电位;(b) MoP@NPC电荷密度图
3.2.2 MoP@NPC氧化脱硫(ODS)性能研究
以* OH为主导的ODS反应有两种活化机制:O-H或O-O键活化机制。作者通过理论计算研究H2O2活化机制。MoP@NPC上H2O2吸附构型有两种:Mo原子与O原子(Type I)或H原子(Type II)的配位(图4)。Type I中H2O2中O-O键被打断,从而在Mo位点表面形成了OH基团的吸附状态,这对* OH形成极为有利;Type II构型中H2O2的O-H键和O-O键被拉断,直接生成H2O。计算表明H2O2更有可能以Type I构型吸附在MoP表面。活性中间产物的解离是ODS反应过程中决定速率的一步。计算纯MoP和NoP@NPC上反应中间体* OH解离能,结果表明,* OH在MoP@NPC上解离能(3.94 eV)比MoP上解离能(5.20 eV)低。
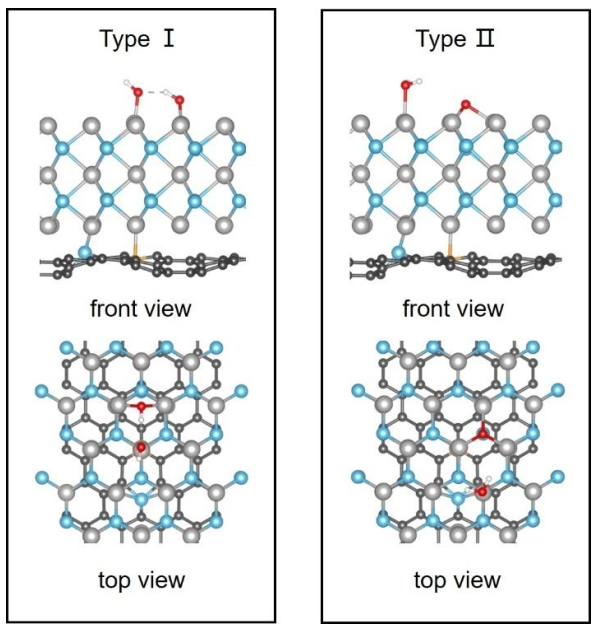
图4 MoP@NPC吸附H2O2两种构型
为了深入理解请EPSI对MoP@NPC催化性能潜在影响。作者计算了参与ODS反应的表面Mo原子态密度(DOS),见图5。MoP@NPC中Mo的d带中心(-0.65 eV)比MoP的d带中心(-0.86 eV)更接近费米能级,表明NPC上MoP具有更高的内在活性。反应中间体* OH解离后,MoP的d带中心上升较大,而MoP@NPC仅轻微移动;表明Mo在解离过程中俘获了* OH的电子。比较发现MoP@NPC上* OH解离所需电子少,更有利于* OH解离和后续氧化。综上所述,强EPSI改变了MoP表面Mo原子电子结构,从而优化了反应中间产物* OH解吸行为,进一步激发了MoP@NPC在氧化噻吩方面的优异催化性能。
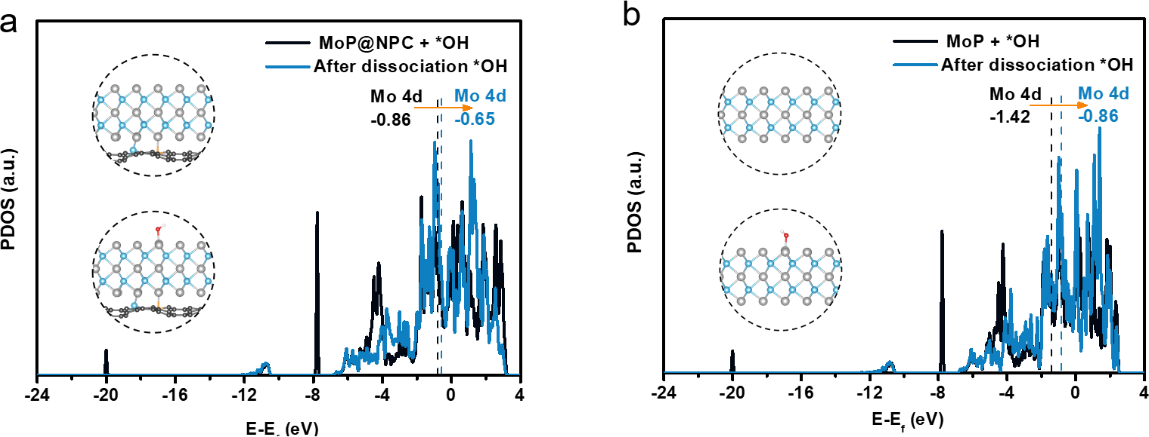
图5 MoP@NPC及MoP吸附*OH的DOS
4. 总结与展望
本案例中,作者在MoP@NPC催化剂中发现了一种新型强EPSI,它与强EMSI和EOSI的概念类似,强EPSI不仅使MoP稳定在低氧化态,还调节其电子结构,从而降低含氧中间产物的解离能,提高氧化脱硫催化活性。MoP@NPC对二苯并噻吩的脱除率高达100%,翻转频率(TOF)值为0.0027 s-1,是不含EPSI的MoP的33倍。这项工作为理解支撑催化剂中的界面相互作用提供一个新的思路,为开发高性能支撑磷化物催化剂开辟新的途径。
参考文献:
DOI: 10.1021/acs.nanolett.3c03217
使用MedeA模块:
MedeA Environment
MedeA VASP


