
一、背景介绍
细胞间质上皮转换因子(c-Met)是受体酪氨酸激酶家族成员,当其与肝细胞生长因子(HGF)结合时,多种细胞信号通路被激活,参与包括增殖、运动、迁移和侵袭等信号通路。在正常生理条件下,c-Met在组织稳态调节中发挥重要作用。据报道,c-Met在不同的人类癌症中通过蛋白质的突变、扩增或过度表达而异常激活。c-Met激酶通过促进细胞散射、侵袭和抗凋亡以及血管生成而帮助癌症扩散。目前,几种用于治疗NSCLC、胃腺癌、肝细胞癌和前列腺癌的口服生物可利用的小分子c-Met抑制剂已被批准用于临床试验。本研究利用分子模拟软件Discovery Studio(简称DS)进行多种计算模拟方法的联合使用,用于发现全新的c-Met抑制剂。
二、研究过程
3D QSAR药效团模型构建与验证
将44个已知c-Met抑制剂分为35个分子的训练集和9个分子的测试集,利用DS模拟软件进行3D QSAR药效团模型构建。在产生药效团模型之前,训练集分子需要先进行预处理。利用DS软件基于CHARMm力场和 polling算法的优质构象模型生成方法生成构象,以确保获得每个化合物的能量最小化构象。根据训练集分子的特征,为该过程选择了四个化学特征:氢键供体(HBD)、氢键受体(HBA)、正电离电位(PI)和芳香环(RA)。不确定度设置为1.5,其他参数使用默认值。
DS的3D QSAR药效团生成协议中使用的HypoGen算法用于识别活性化合物的共同特征,同时排除构象允许空间区域内非活性化合物的特征。根据Cost值和统计参数,共输出了排名最高的十个药效团模型。好的药效团模型应具有较高的相关系数、最低的Total cost值和最低的RMSD值。Total cost值应接近Fixed cost,远离Null cost值。Null cost 与 Total cost的差值表明药效团模型的可靠性。表1展示了前十个药效团模型及其统计参数。优质药效团模型Hypo1(图1)具有优质相关系数(0.8798)、最高Cost差值(357.921)和最低RMSD(2.49Å),模型中包括了氢键受体(HBA)、疏水性(HYP)、正可电离(PI)和环芳香(RA)特征。对于Cost分析,使用了两个关键值:Fixed cost和Null cost之间的差值以及Null cost和Total cost之间的差值。357.921的Cost差值表明Hypo 1具有90%以上的统计显著性,因此选择Hypo 1作为进一步虚拟筛选的优质药效团模型。
表1:输出的前10个3D QSAR药效团模型参数
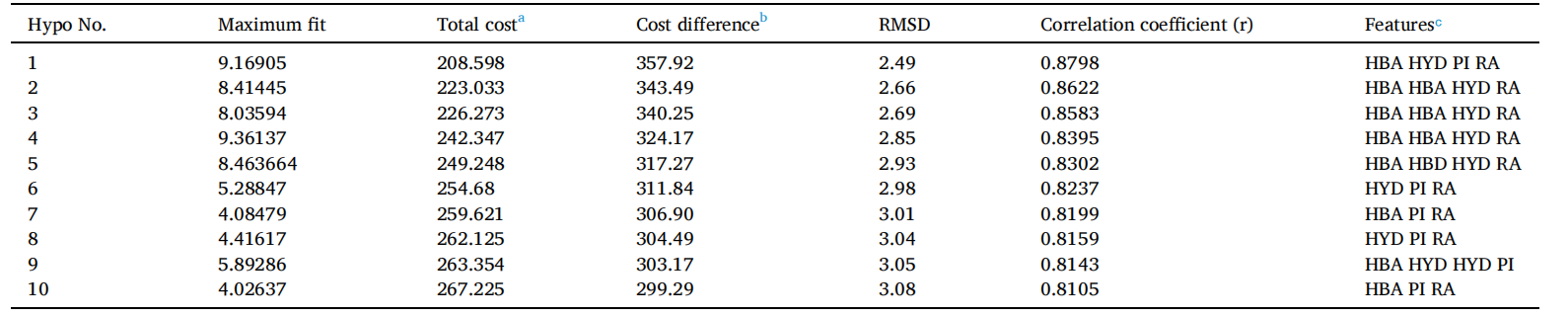

图1:Hypo1药效团模型
优质药效团模型Hypo1进一步通过测试集进一步评估。表2中展示了训练集分子的实验和预测活性。除此之外,通过将正在临床试验的II类c-Met抑制剂与Hypo1进行匹配和活性预测,进一步评估药效团模型(图2和表3)。
表2:测试集分子IC50实验值与预测值比较


图2:II类c-Met抑制剂与Hypo1的匹配情况
表3:II类c-Met抑制剂的化合物的药效团模型拟合值、对接得分及其实验和估计IC50值

骨架跃迁法构建分子库
先导化合物优化是一个复杂的迭代过程,旨在优化有前景的化合物的化学结构,以提高生物活性、选择性、生物利用度、药代动力学、药效学参数,并降低毒性,从而发现临床前候选药物。本文利用已报到的c-Met抑制剂1FN为先导化合物,利用DS中的Replace Fragment功能模块从默认片段库中识别并自动替换初始分子中指定的片段,共生成了815个类似物的分子库。
虚拟筛选
利用Hypo1模型对Drug-LikeDiversity、MiniMaybridge、scPDB以及骨架跃迁产生的分子库进行虚拟筛选。结果显示骨架跃迁产生的分子库中仅四个分子的Fit value值大于1FN(表4),而其它数据库中也同样具有四个分子的Fit value值大于1FN(表5)。
表4:先导化合物1FN与骨架跃迁数据库虚拟筛选的4个候选分子的模拟计算结果

表5:1FN和cabozantinib与虚拟筛选4个候选分子的模拟计算结果

分子对接
除了药效团模型之外,分子对接也是常用于先导化合物发现和优化的计算模拟程序。DS中的分子对接程序CDOCKER是基于CHARMm的半柔性对接程序,采用模拟退火进行能量优化,从而使对接结果更加准确。对1FN和虚拟筛选的8个候选分子进行CDOCKER对接(表4.表5),结果显示,除3CE3 81外,其余7个分子的-CDOCKER energy大于1FN,表明其生物活性可能优于1FN。
结合自由能计算
分子力学的势能包括键合和非键合相互作用(范德华力和静电相互作用能),而溶剂化的自由能包括极性溶剂化能(静电)和非极性溶剂解能(非静电)。使用DS软件通过MM-PBSA进行结合自由能计算,一般来说,结合自由能越小,复合物越稳定,配体和受体之间的结合越强。表6总结了所有配合物的相互作用能和结合自由能。可以看出虚拟筛选的8个候选分子中,除了CBG98066,其余分子的结合自由能与1FN相近且优于Cabozantinib,进一步证明了其抑制c-Met的潜力。
表6:MM-PBSA自由能计算结果

分子动力学模拟
分子动力学是一门结合数学、物理和化学的综合技术,该方法主要是依靠牛顿力学来模拟分子体系的运动,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。使用DS进行分子动力学模拟,通过在预定时间段内保持温度、体积和压力参数恒定,可以确定复合物中原子的运动,研究复合物的稳定性。结果显示,除了CBG98066,在整个模拟期间,系统基本稳定,结构没有重大变化(图3),表明虚拟筛选的7个候选分子都与c-Met的活性口袋紧密结合,其中化合物3CE365具有最小的RMSD平均值,似乎比1FN表现出更好的主链稳定性。相比较RMSD,RMSF值被认为是动力学模拟中整体灵活性的标准,如图4所示,1FN复合物的RMSF平均值为0.834,所有命中的RMSF值范围为0.797至0.867,化合物3CE381的RMSF最小平均值为0.797。上述分子动力学结果表明,除了CBG98066,其余7个候选分子能够与c-Met形成稳定的复合物,具有抑制c-Met活性的潜力。

图3:分子动力学模拟RMSD结果图

图4:分子动力学模拟RMSF结果图
三、结语
本研究中,利用分子模拟软件DS发现全新的c-Met激酶抑制剂。首先通过已知c-Met激酶抑制剂构建并验证具有活性预测能力的3D QSAR药效团模型,用于筛选c-Met激酶抑制剂。结果分析表明,c-Met激酶抑制剂药效团模型由四个药效团特征组成:一个氢键受体(HBA)、一个芳香环(RA)、一种正离子特征(PI)和一个疏水特征(HYD)。优质药效团模型Hypo1具有优质相关系数(0.8798)、最高Cost差值(357.921)和最低RMSD(2.49Å)。除了测试集之外,还利用已报道的II类c-Met抑制剂对药效团模型进行外部验证。所有结果表明,该模型能够可靠地预测c-Met酪氨酸激酶的抑制剂的活性。随后Hypo 1被进一步用于筛选三个小分子数据库以及基于先导化合物1FN通过骨架跃迁策略产生的配体数据库。虚拟筛选得到的8个候选分子,进一步通过分子对接验证。分子对接获得的蛋白配体复合物,再进行分子动力学模拟,结果表明,除了CBG98066,剩余七个小分子都具有良好的抑制c-Met潜力,值得进一步的生物活性评估。
参考文献
Asmaa Raafat,Samar Mowafy,Sahar M. Abouseri,Marwa A. Fouad,Nahla A.Farag,Lead generation of cysteine based mesenchymal epithelial transition (c-Met) kinase inhibitors:Using structure-based scaffold hopping,3D-QSAR pharmacophore modeling,virtual screening, molecular docking,and molecular dynamics simulation,Computers in Biology and Medicine,Volume 146,2022,105526,https://doi.org/10.1016/j.compbiomed.2022.105526.


