
多相Co-N-C分子催化剂高效电化学合成H2O2
关键词:电催化、电子ORR、H2O2合成、高选择性
1. 案例背景
过氧化氢是一种重要的氧化剂,广泛用于化学工业、环境保护和医疗卫生领域。钴卟啉β-取代基/碳纳米管催化剂能利用双电子氧还原反应(2e--ORR)在0.1M HClO4电解质中显著提高H2O2的产量和选择性。经实验证明,将卟啉β位的8个氢原子用氟取代后(即CoPorF/CNT),再吸附到碳纳米管上,可以协同调控催化剂的活性,达到最佳催化效果。在200 mV的过电压下,H2O2的选择性超过94%,每秒转换频率(TOF)达到3.51 s-1;在双电极电解槽中,最大H2O2产率为10.76 molH2O2·gcat-1·h-1,同时能有效抑制副反应(四电子氧还原反应)的发生。
2. 建模与计算方法
作者在MedeA Environment中构建了四种不同结构的钴卟啉化合物CoPorX(X=H、Et、Br和F)。使用Builder Nanotubes功能创建了C纳米管;将四种CoPorX分别吸附到碳纳米管(CNT)上后,成功构建了非均相催化剂CoPorX/CNT。作者采用MedeA-VASP模块中GGA-PBE方法,对体系进行结构优化并计算了差分电荷密度、*O2结合能、*HOOH结合能等性质,并计算得到了沿不同反应路径的理论ORR电位和反应选择性。结构优化时,模型被完全放开,力收敛精度为0.05 eV/Å。
3. 结果与讨论
3.1 理论预测
钴卟啉是电催化合成H2O2的良好催化剂,其结构如图1a所示。为设计性能更加优异的催化剂,可以通过调控Co活性中心与ORR中间产物至最佳结合强度得到。钴卟啉上有meso-位和β-位两个官能团位点,改变不同位点上的官能,团利用位阻效应和辅助因子(co-factor)效应改变结合强度,能设计出性能更优异的电催化剂。
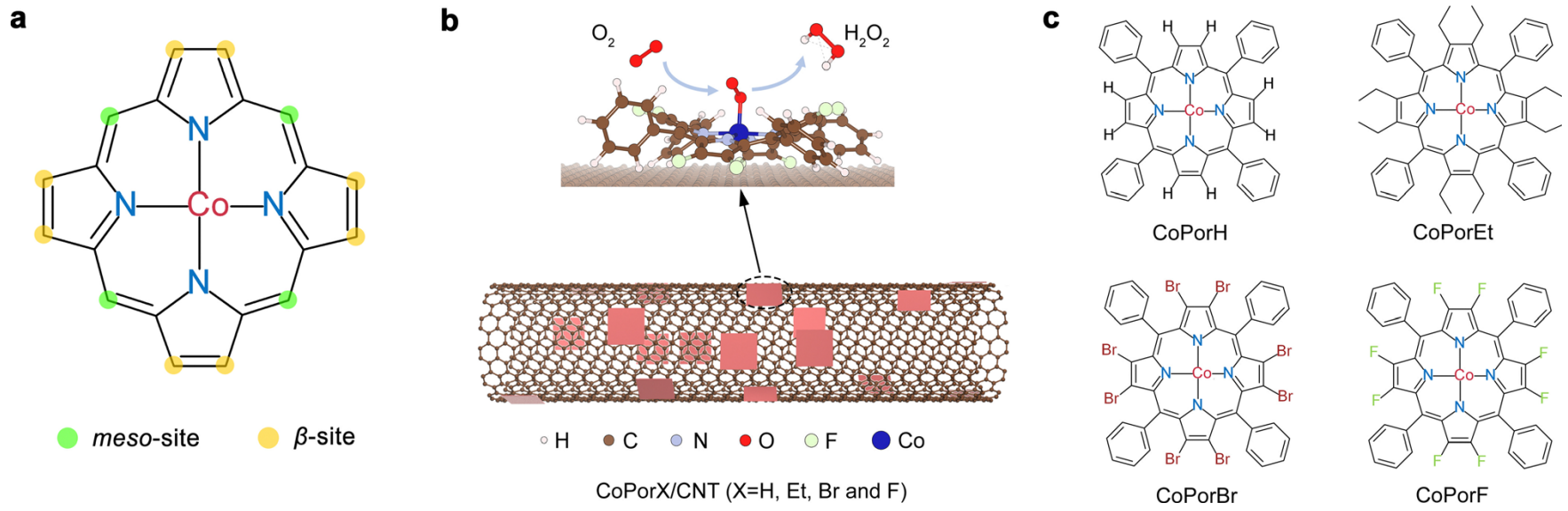
图1 (a) 卟啉化学结构和两种取代位;(b) ORR电合成原理及催化剂结构示意图;(c) 本文合成的钴卟啉分子结构
作者在MedeA Environment中构建了四种钴卟啉化合物CoPorX(X=H、Et、Br和F),并利用范德华力将它们吸附到碳纳米管(CNT)上,形成了CoPorX/CNT非均相催化剂(图1b-c)。通过MedeA VASP计算了这些化合物的电子结构及其与ORR中间产物的结合能。结果显示,CoPorH/CNT和CoPorF/CNT中的Co原子带正电性,而CoPorBr/CNT和CoPorEt/CNT中则带负电性(见图2a)。进一步分析了Co活性中心与不同ORR中间体的相互作用,发现了它们之间的结合能与催化活性的关系,特别是Et取代后的CoPorEt/CNT与O2中间体的结合能最弱,而F和Br取代则最强(见图2b、2c)。这些结果揭示了在设计催化剂时,Co活性中心与O2和*HOOH中间体的吸附强度需适中,才能实现高效的反应转化。
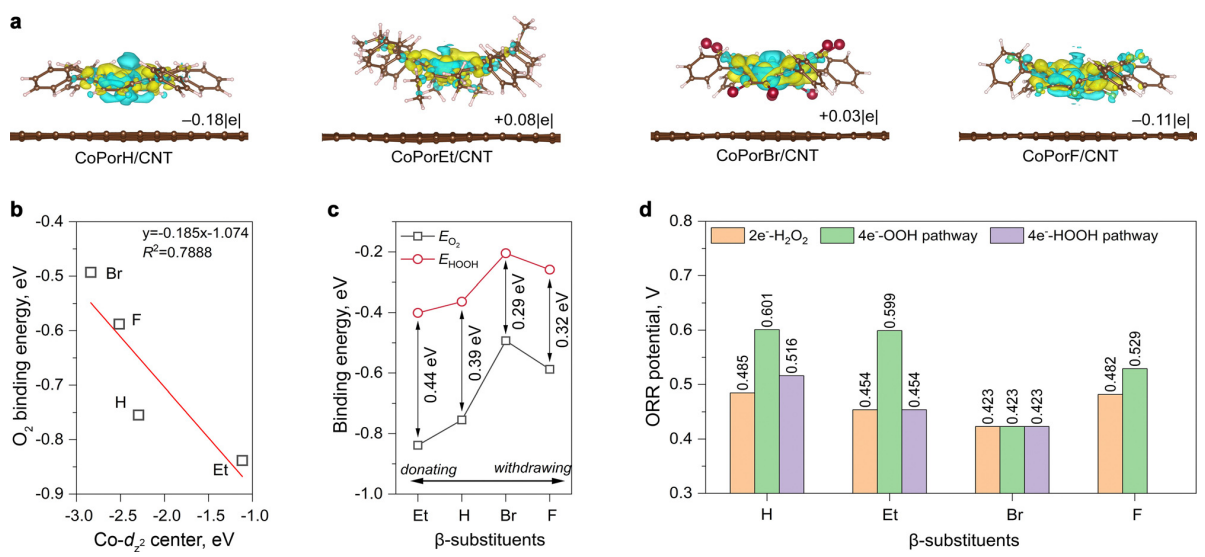
图2 (a) 不同CoPorX催化剂的电荷密度(黄色和青色分别代表电荷积累和消耗);(b) 不同CoPorX催化剂的Co-dz2轨道中心与*O2结合能之间的关系;(c) 不同CoPorX催化剂与*O2和*HOOH中间体结合能之间的关系;(d) 沿不同反应路径的理论ORR电位。
比较了不同CoPorX催化剂的理论电势,如图2d所示,发现H、F取代的催化剂,其电势分别为0.485和0.482 V,优于Et和Br的0.454和0.423 V,但伴随着副反应4e—ORR的发生。而吸电子的F和Br取代的催化剂,几乎不会催化副反应,特别是CoPorF催化剂。预测的ORR反应活性顺序为H≈F>Et>Br,H2O2选择性顺序为Br>F>H>Et。
3.2 催化剂实验表征
在理论计算的指导下,作者合成了四种结构的催化剂,并进行了表征。发现四种催化剂的带隙,及其在Co金属化前后的变化与计算结果非常吻合(图3a)。钴卟啉分子在CNT载体上高度分散,没有形成聚集体(图3b)。在X射线近边吸收光谱(XANES)中,Co在卟啉核中呈现D4h对称性,放大边缘区域后,发现不同β取代的卟啉中的Co价态接近2+(图3c-d)。同时,Co在Co–N键长和卟啉核畸变参数之间建立了良好的相关性(图3e),因此卟啉核变形主要是由鞍形变形所致,取代基体积越大,形变越显著。

图3 (a) 不同CoPorX催化剂的UV光谱带隙;(b) 催化剂的HAADF-STEM图,从左起依次为CoPorH/CNT、CoPorEt/CNT、CoPor Br/CNT和CoPorF/CNT;(c) Co的K-edge XANES谱图;(d) 边缘区域放大图;(e) Co–N键长与卟啉分子变形参数之间的关系;(f) 卟啉分子在CNT上吸附前后的高分辨率Co2p XPS谱图;(g) 在2p3/2轨道上Co2p的XPS光谱差异。
在Co2p XPS谱图中,四种钴卟啉在大约780和795 eV处显示出自旋轨道分裂的2p3/2和2p1/2峰,在CNT上吸附后,所有化合物出现了0.1–0.2 eV的位移,CoPorH/CNT和CoPorF/CN的2pz峰移向更高的结合能,而CoPorEt/CNT和CoPorBr/CNT则呈相反趋势,证实了吸附的钴卟啉分子与CNT之间强烈的电子相互作用(图3f-g)。
3.3 H2O2活性测试
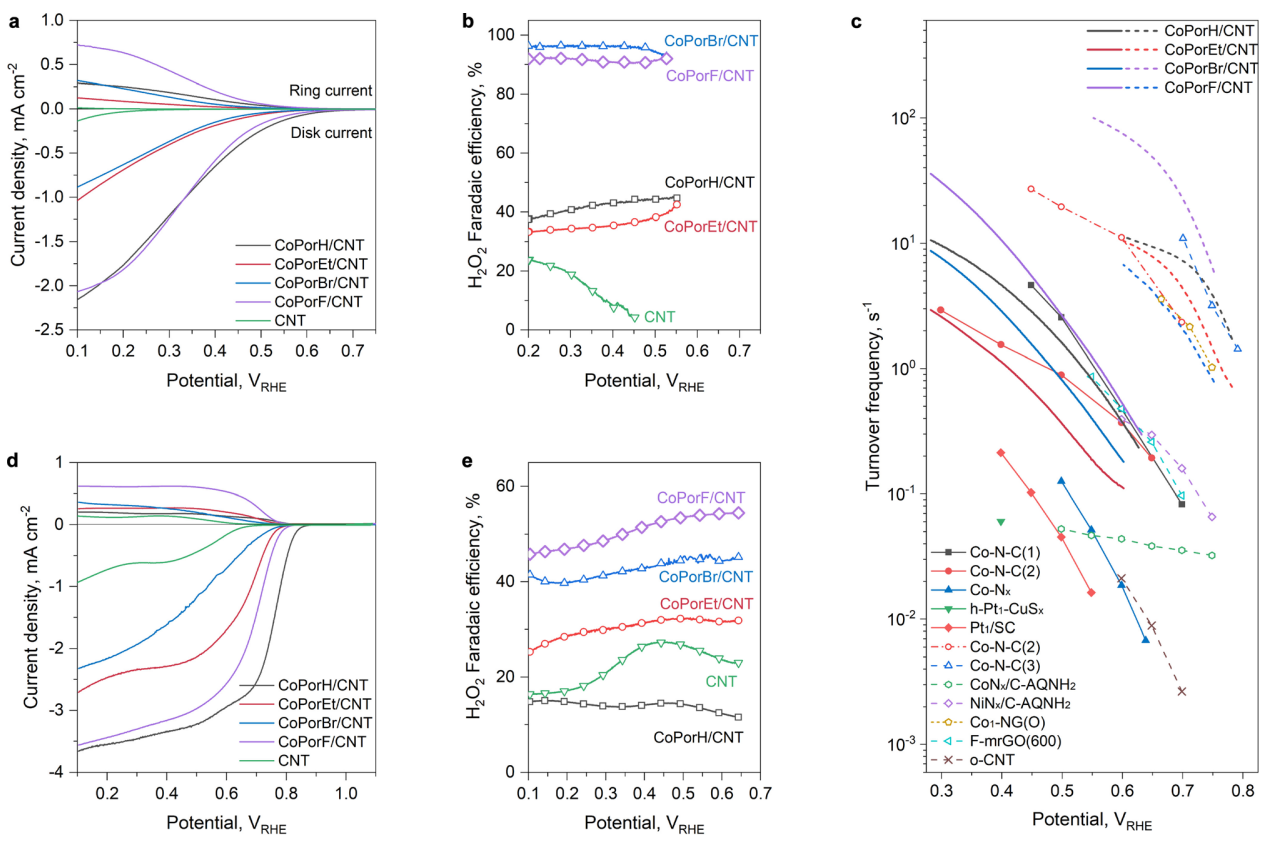
图4 (a) LSV曲线和 (b) 在O2饱和0.1 M HClO4电解质中的H2O2法拉第效率(扫描速率:5 mV·s-1,转速:1600 rpm);(c) 与文献报道的催化剂做比较;(d) LSV曲线和 (e) 在O2饱和0.1M KOH电解质中的H2O2法拉第效率。
线性扫描伏安测试(LSV)。在酸性条件下,CoPorF/CNT和CoPorH/CNT表现出较小的起始电位差,并且CoPorF/CNT在200 mV过电势下的H2O2转化效率最高,为89.3%。通过比较H2O2每秒转换频率(TOF)和总质量活性,的TOF达到3.51±0.06 s-1,总质量活性和比质量活性分别达到22.01 A·gcat-1和11584 A·gCo-1,高于其它催化剂。在碱性条件下,四种催化剂显示出更高的电流密度,但H2O2选择性有所下降,其中CoPorF/CNT的性能仍然最佳,表现出作为环境友好型H2O2生产的有效催化剂潜力。
4. 总结与展望
作者设计了一种钴卟啉β-取代基/碳纳米管结构催化剂,用于高效电化学合成H2O2。理论计算表明,β-位取代基能有效调控碳纳米管上钴活性中心的电子性质和催化活性,尤其是氟取代的CoPorF表现最佳。此外,β-位取代还能通过影响卟啉构象提高催化剂的稳定性。CoPorF/CNT催化剂在酸性和碱性电解质中展示出高周转频率(分别为3.51和85.1 s-1),其质量活性达10.76 mol·gcat-1·h-1,能够生产纯净的H2O2水溶液,适用于水处理和化学合成。本案例为理解单原子活性中心在催化性能中的作用提供了重要见解,为未来其他反应的催化剂设计提供了新思路。


