
【机器学习力场培训班】
VASP的从头算分子动力学(AIMD)模拟功能目前在各领域基础研究中被广泛应用,源于其能够准确地预测材料微观结构的演化过程。但对于较大的体系,AIMD的耗时相对较长,对很多科研工作者而言都是一个难题。VASP6版本的一个重要的功能——实时机器学习力场(MLFF)能够解决这一难题,能大幅加速分子动力学模拟。因此,自从此功能推出以来,始终具有很高的关注度。
为了让更多用户了解全新的MLFF功能,源资科技于7月30日举办VASP6机器学习力场专题速成班,并对课程进行了精简升级,更新了课程讲义和上机操作手册。全程免费,包含授课及上机指导。
培训时间虽然仅有半天,但是学员们收获满满:不但了解到机器学习力场的理论基础和实际应用案例,而且通过上机实操掌握了大量建模和计算技巧。讲师与学员积极互动交流,回答大量学员关心的MLFF相关问题,获得了一致好评!
由于本次培训参与人数较多,学员积极谈论,反响热烈,小编也收集了很多问题,由于篇幅有限,我们分成了三期Q&A完成答疑分享。
【Q&A答疑分享——Part 2】
1. 两个固相的界面跑AIMD是使用NPT还是NVT系综?
A:用NVT系综。
2. 老师我想请问如果我想跑同一个体系 50ps的AIMD(同一个温度压强不考虑相变),我是直接用 MLFF 跑 50 ps,收集数据;还是先训练出一个合理的力场,再用力场跑更久的?
A:推荐使用后者,能保证整段分子动力学的误差是前后一致的。
3. 误差多少可以认为力场是合理的?
A:能量的均方根误差和贝叶斯误差小于5meV/atom,力的均方根误差和贝叶斯误差均小于0.1eV/Ang。
4. 是否可以用原胞训练获得的力场来跑超胞的分子动力学?
A:可以选取适当大小的晶胞(推荐十几到几十原子这个大小)进行MLFF的训练,然后将得到的力场用于超胞分子动力学模拟。
5. 产生训练集过程中,训练的步长和时间一般要多久呢?
A:步长需要根据体系决定,总模拟时长需要经过测试,一般而言贝叶斯误差和均方根误差达到一个合理的程度(参考Q3),就可以认为当前训练时间合理。
6. 上机用的.flow文件是什么?
A:是MedeA中的流程图文件,可以直接导入和使用,也可以保存后共享给其他人,非常方便。
7. 我想自己对贝叶斯误差作图,需要找哪个文件?
A:可以找Stage_1中的MLFF_BayesianErrors.dat文件,里面包含了详细的贝叶斯误差。

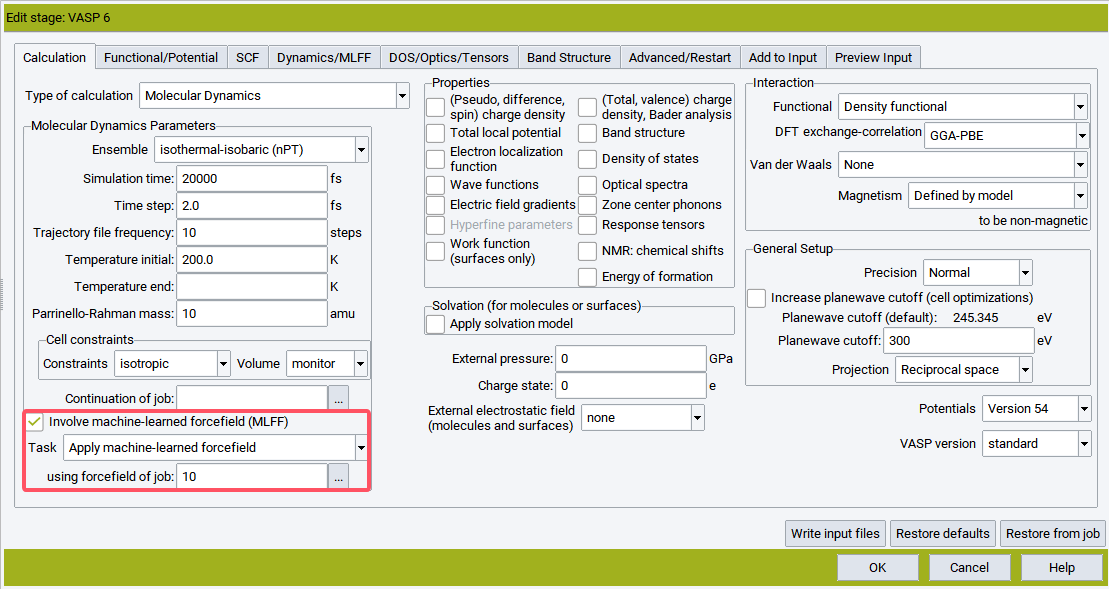
8. 训练完得到的力场如何使用?
A:在VASP中,需要将训练完得到的ML_FFN拷贝到新作业的文件夹中,并命名为ML_FF,将ML_ISTART设置为2。在MedeA中,需要将Task设置为Apply machine-learned forcefield,并在using forcefield of job之后填写训练力场对应的作业序号。
9. AIMD温度波动很大对训练有影响吗?
A:没有太大影响,但如果温度波动过大导致偏离目标结构,会导致无法获得预期的力场训练结果。
10. 我看网上说在训练时刚开始的数据集是不大准确的,所以例如在某个温度下先跑个1ps,然后再用1ps之后的结构作为初始结构再重新训练力场,这样的力场才比较准确,这种说法正确吗?
A:可以这样做,思路没有问题。


